
Dr. Alessandro Giardini
If you've been told your child may have Marfan syndrome, it's completely understandable to feel worried. Most parents immediately think about the heart, and especially the aorta.
The important thing to know is this: with the right monitoring and care, most children with Marfan syndrome do very well. Life expectancy today, with early diagnosis and appropriate management, can sit close to that of the general population.
Marfan syndrome is a genetic condition that affects connective tissue, the material that holds the body together and gives strength and flexibility to blood vessels, bones, ligaments, muscles and skin. The condition takes its name from Antoine Marfan, a French doctor who first described it in 1896. It ranks among the most common inherited connective tissue disorders, affecting roughly 1 in 3,000 to 5,000 people worldwide, and occurs equally in boys and girls.
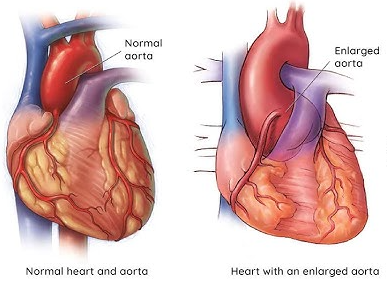
Because connective tissue runs throughout the body, Marfan syndrome can involve many different systems, including the heart and blood vessels, the skeleton, the eyes, the lungs and the skin. In children, the most important area we monitor is the aorta, the main blood vessel carrying blood away from the heart.
A change (mutation) in a gene called FBN1 on chromosome 15 causes Marfan syndrome. This gene provides the instructions for making a protein called fibrillin-1, one of the main building blocks of the tiny fibres that give connective tissue its strength and elasticity. The mutation also drives increased activity of a signalling system called the TGF-beta pathway, which contributes to many of the features of the condition, particularly the widening of the aorta.
Marfan syndrome follows an autosomal dominant pattern of inheritance. If a parent has Marfan syndrome, each child has a 1 in 2 (50 per cent) chance of inheriting it. About 75 per cent of cases are inherited from a parent, though in some cases nobody has yet diagnosed the parent. In the remaining 25 per cent, the gene change occurs for the first time in the child with no family history. The condition varies considerably from person to person, and even two people in the same family carrying the same gene change may experience it very differently.
The aorta, particularly the section closest to the heart called the aortic root, tends to widen gradually over time in children with Marfan syndrome. Without monitoring and management, this widening can lead to an aortic aneurysm (a dangerous bulge in the wall) or, in serious cases, an aortic dissection (a tear in the wall), both of which constitute medical emergencies.
The mitral valve, which sits between the two left chambers of the heart, also commonly develops problems. Mitral valve prolapse, where the valve flaps become floppy and do not close properly, occurs in many children with Marfan syndrome and can sometimes cause the valve to leak.
The good news is that we can detect changes early with echocardiography, slow progression with medication when needed, and plan treatment safely and well in advance. This is why early assessment and regular follow-up are essential.
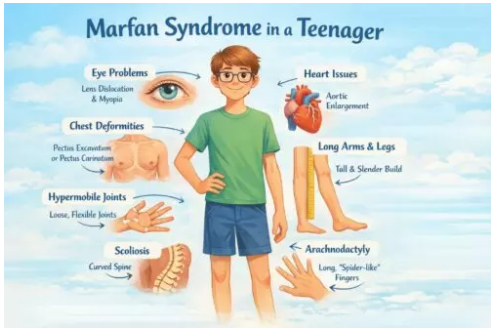
Many features of Marfan syndrome start mild and become more apparent during the teenage growth spurt, which means doctors can find the condition hard to diagnose in younger children. Not every child will have all of these features.
Common signs include a tall, slim build with long arms, legs, fingers and toes (doctors describe the long fingers as arachnodactyly). Other features can include a sunken chest (pectus excavatum) or a protruding chest (pectus carinatum), curvature of the spine (scoliosis), very flexible joints, flat feet and a high-arched palate that may crowd the teeth.
Eye problems form a hallmark of Marfan syndrome and one of the features that help doctors make the diagnosis. The most characteristic finding is dislocation of the lens of the eye (ectopia lentis), which occurs in about 60 per cent of people with Marfan syndrome. Short-sightedness is very common, often more severe than usual. Children also face an increased risk of retinal detachment, glaucoma and early cataracts.
Some children have very subtle signs. Others are identified because of a family history. If a family history exists, doctors can monitor children from birth. If there is no family history, the diagnosis may not come until a child is older.
.png)
Diagnosing Marfan syndrome involves bringing together information from a physical examination, heart scans, eye tests, family history and genetic testing. Doctors use internationally agreed criteria known as the revised Ghent nosology to reach the diagnosis.
A specialist carries out a detailed examination looking at height, body proportions, chest shape, spine, joints and facial features. An echocardiogram measures the size of the aortic root, checks the heart valves and assesses how well the heart pumps. This stands as the single most important test in monitoring Marfan syndrome. A thorough eye examination by an ophthalmologist checks for lens dislocation, short-sightedness and retinal problems.
Genetic testing through a blood test identifies changes in the FBN1 gene in about 95 per cent of children with Marfan syndrome. This helps confirm the diagnosis and allows testing of other family members. CT or MRI scans may look at the full length of the aorta and other blood vessels, particularly in older children and adolescents. Doctors generally prefer MRI in children because it avoids radiation.
Because 75 per cent of cases run in families, first-degree relatives of a diagnosed child should receive an assessment. This can involve a clinical examination, an echocardiogram and genetic testing. Sometimes a parent discovers they have the condition only after their child receives a diagnosis. Finding affected family members early is one of the most valuable steps a family can take.
No cure for Marfan syndrome exists, but it ranks among the most treatable genetic conditions. With the right management, the vast majority of children go on to live long, active and fulfilling lives.
Medication. Beta-blockers (such as atenolol) are the most commonly used drugs, slowing the heart rate and lowering blood pressure to reduce the force on the aortic wall. Angiotensin receptor blockers (ARBs), particularly losartan, are also widely used. Research has shown that losartan directly affects the TGF-beta signalling pathway that drives Marfan syndrome, and it may offer additional protection beyond blood pressure lowering alone. Some children take a combination of both, particularly if the aorta is enlarging more quickly than expected.
Regular monitoring. Most children have an echocardiogram at least once a year. If the aorta is getting bigger or there are other concerns, the team may arrange scans more frequently. Eye examinations should take place regularly throughout childhood. The team should check the spine at each visit, particularly during puberty, as scoliosis can progress quickly. I work with each family to develop a monitoring plan that is thorough but also practical.
Surgery. Doctors recommend preventive surgery when the aortic root reaches a size at which the risk of a tear becomes significant, typically 5.0 cm, though the team may consider surgery earlier (at 4.5 cm) if additional risk factors exist. The most commonly performed operation is a valve-sparing aortic root replacement, where the surgeon replaces the weakened section of the aorta with a synthetic tube while keeping the patient's own aortic valve. Planned surgery carries a very low risk at experienced centres.
Exercise and activity. Staying active matters for children with Marfan syndrome, both for physical health and emotional wellbeing. Activities such as swimming, cycling, walking, golf, doubles tennis and gentle hiking are generally safe. The key is that your child should feel comfortable enough to hold a conversation while doing the activity. Families should steer children away from competitive and contact sports, heavy weight-lifting and high-intensity endurance activities. As a specialist with a particular interest in exercise physiology in children with heart conditions, I help families navigate these decisions so that children can enjoy staying active while remaining within safe limits.
Eyes, bones and joints. Glasses or contact lenses can correct short-sightedness, and surgery may be needed if the lens dislocates significantly. Scoliosis may require bracing or surgery. Supportive insoles can manage flat feet. Dental crowding may need orthodontic treatment. Physiotherapy can help with joint pain and hypermobility.
Emotional and psychological support. Living with a lifelong condition that affects appearance and limits some activities can feel challenging, particularly during the teenage years. School staff should know about the condition. Connecting with other young people through organisations like the Marfan Foundation or the Marfan Trust can make a real difference.
In most cases, yes. With appropriate care, children attend school normally, physical activity is encouraged with guidance, and the long-term outlook is very good. Before modern treatments existed, life expectancy fell significantly short of normal. Today, with early diagnosis and proper management, life expectancy has improved enormously. The key is structured, specialist follow-up.
You should seek review if your child has been diagnosed or suspected of having Marfan syndrome, there is a family history, you've been told there may be aortic enlargement, or you simply want reassurance.
Yes. Some children have very subtle features and may only need monitoring. The condition varies considerably from person to person.
It can be diagnosed at any age, though many features become more noticeable during the teenage growth spurt. If a family history exists, doctors can monitor children from birth.
Usually slowly, which is why regular monitoring with echocardiography is effective. If the aorta is enlarging more quickly than expected, the team will increase the frequency of scans and may adjust medication.
Yes, but some high-intensity or contact sports may need to be limited depending on the aorta. Swimming, cycling, walking, golf and doubles tennis are generally safe. Your child's cardiologist will give personalised guidance.
Yes, it usually runs in families with a 50 per cent chance of passing it on. In about 25 per cent of cases, it occurs for the first time with no family history.
Most children do not need surgery if monitored appropriately. When surgery is recommended, it is typically a planned, preventive procedure with an excellent safety record.
Most children have an echocardiogram at least once a year. If there are concerns about the rate of aortic growth, scans may be arranged every six months or more frequently.
Yes. Parents and siblings of a diagnosed child should strongly consider assessment, including a clinical examination, echocardiogram and genetic testing if the family's gene change is known.
The two conditions share some features, especially widening of the aorta. However, lens dislocation is a hallmark of Marfan syndrome but does not occur in Loeys-Dietz syndrome. Conversely, Loeys-Dietz syndrome features widely spaced eyes, a split uvula and twisted arteries. In Loeys-Dietz syndrome, the aorta can tear at smaller sizes, so doctors recommend surgery at a lower threshold. Different genes cause the two conditions, and each requires a slightly different approach to monitoring and treatment.
Sudden severe pain in the chest, back or abdomen, especially if it feels like a tearing or ripping sensation, may signal an aortic dissection. Call 999 immediately. Your child should carry a medical alert card or bracelet stating that they have Marfan syndrome.
Author: Dr. Alessandro Giardini, MD, PhD
Written 30/03/2026